
Electronic Trial Master File (eTMF)
Document Management System for Clinical Studies
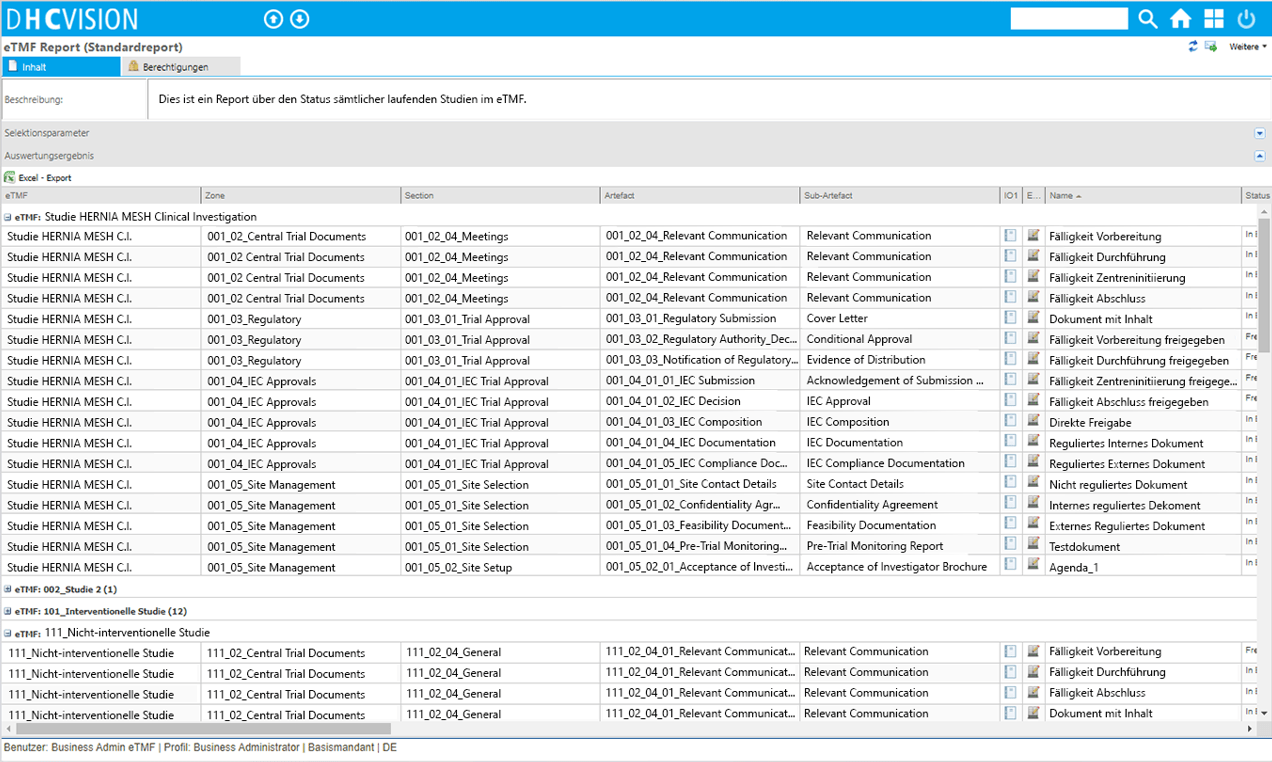
Complex structures and distributed processes are characteristic of clinical trials. Especially the documentation of activities, events, and results in the Trial Master File (TMF) is a challenge: It involves a high number of very heterogeneous documents. The DHC VISION eTMF solution supports reliable structuring and classification of the entire documentation according to Good Clinical Practice (GCP).
A clinical trial tests the performance, efficacy and safety of medical devices, forms of treatment, active substances, etc. and is conducted with patients or volunteers. GCP – Good Clinical Practice – defines an internationally recognised standard for planning, conducting, monitoring/auditing, documenting, evaluating and reporting clinical trials, based on ethical and scientific aspects.
The Trial Master File (TMF) is a collection of documents from a clinical trial. All essential files of the trial are summarised in the TMF. The TMF ensures the integrity of the clinical data and compliance with GCP. However, audits repeatedly find serious weaknesses in the regulatory-compliant and traceable documentation of studies: the TMF is incomplete, not always up to date, and documents are not consolidated in one place at the end of the study. Delays in the approval of active substances and medical devices or even a failure of studies are the frequent consequences.
Impressive functionality
Study management
- Digital document management according to the Drug Information Association (DIA) reference model
- Template-based creation and flexible configuration of studies
- Easy structuring of TMFs for studies
- Differentiated role and authorization concept for studies and study documents
- Flexible control of access to storage structures and individual documents
- Clear and auditable filing of documents in individual studies
- Convenient import and simple classification of documents
- Secure creation, review and approval of documents in the system
- Availability of standard document types according to DIA reference model


Document Management
- Standardization based on more than 50 document templates and workflows
- Flexible document encoding
- Electronic signatures compliant with 21 CFR Part 11
- Status management
- Comment function and document in workflow for communication
- Automated calculation of validity and archive duration
- Target-group specific distribution for new / changed documents
- Distribution of documents for information or acknowledgement (persons, groups)
- Messages sent by mail resp. to the personal task box
- Real-time publication
- Simple and flexible adaptation of release workflows
- Distribution of tasks to the personal task box as well as by e-mail
- Systematic procedures for invalidation, validity extension, reactivation and archiving of documents
Document Control
- Comprehensive monitoring reports
- Full traceability and control in document management
- Consistent change and version history of document
- Highly performant cognitive search
- Comprehensive search mechanisms with filtering/selection functions
- Reminder/escalation mechanisms
- Comprehensive status tracking (tracebility)
- Workflow-based status monitoring
- Interactive data analytics and business intelligence functions
- Diagnosis of current and future compliance status

Request factsheet
Compact information on all processes related to controlled documents and the complete range of functions are available in the factsheet on DHC VISION Electronic Trial Master File.
"*" indicates required fields
All options at a glance
Study and document management
- Full implementation of the DIA reference model
- Template-based creation and configuration of studies based on the DIA reference model
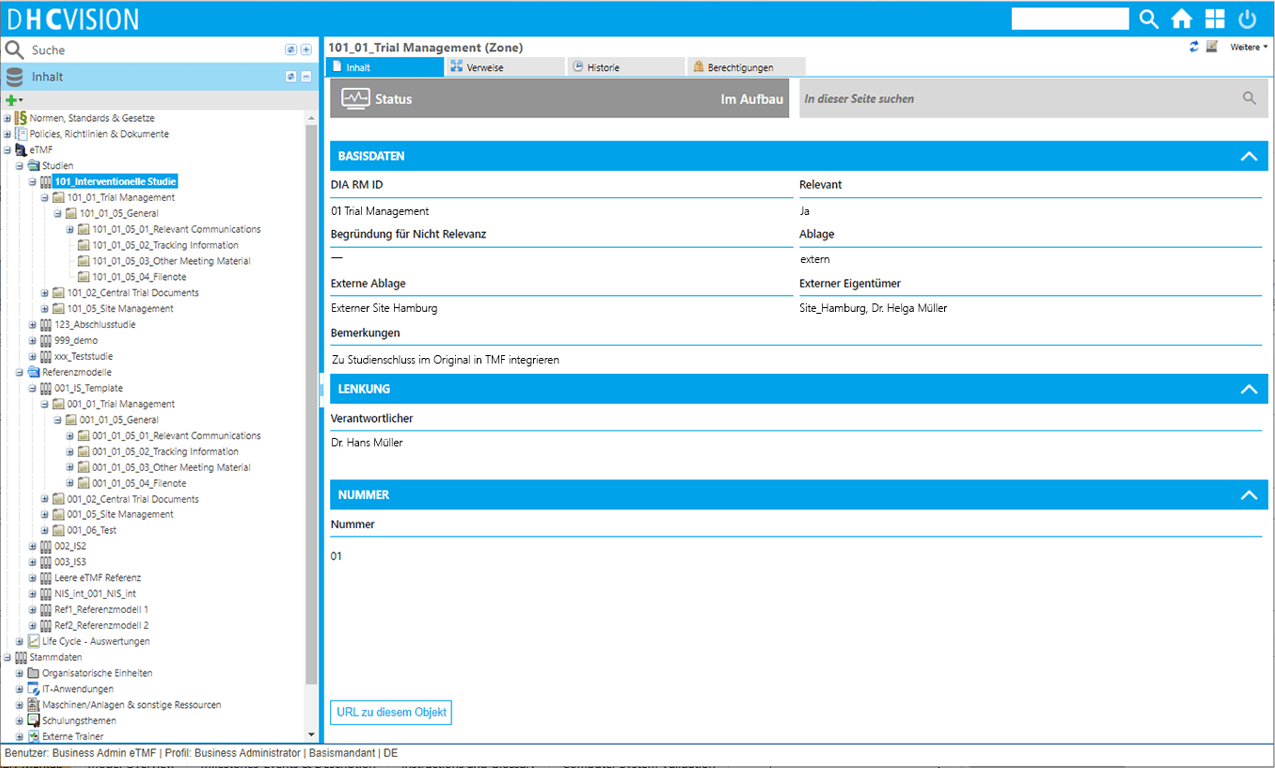
- Management and tracking of externally stored (e.g. at sites) documents
- Easy structuring of TMFs for individual studies
- Availability of standard study document types
- Flexible control of access to file structures and individual documents
- Clear and auditable storage of documents in individual studies
- Import, migration, and classification of documents
- Automated assignment of unique document numbers (prefix)
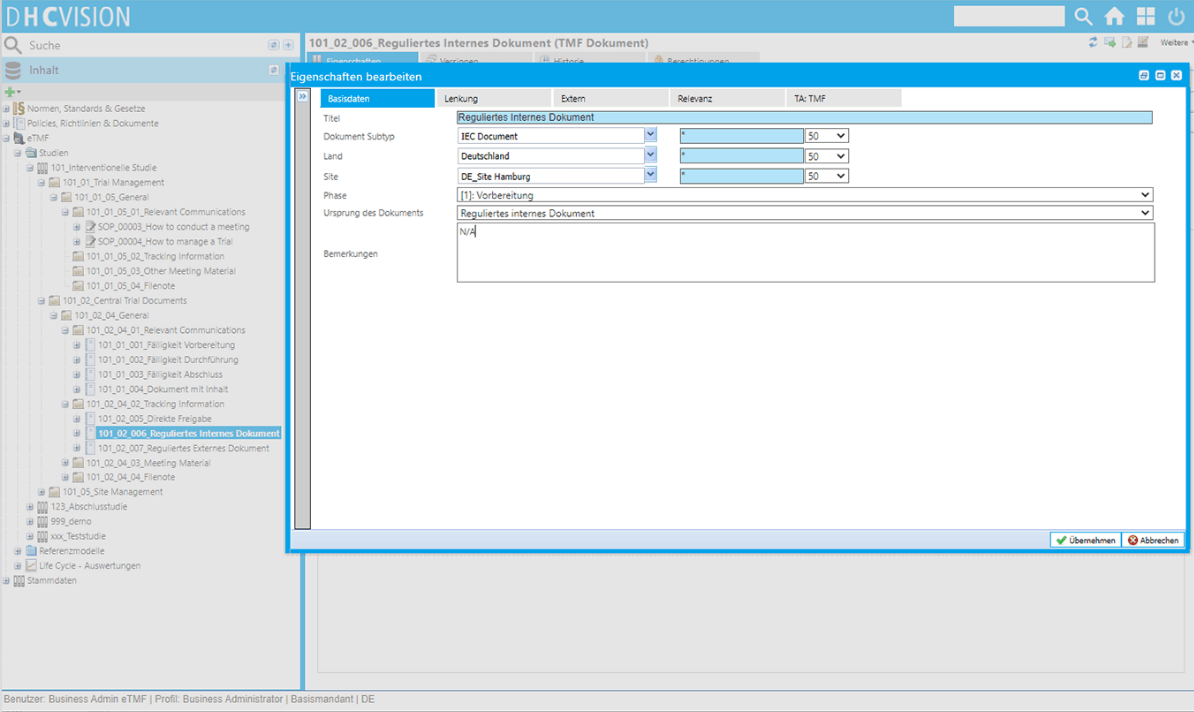
- Flexible coding of documents as well as freely definable properties or attributes
- Automated calculation of revision cycles and archive duration
- Import of study documents
- Integration of Microsoft Office products (Word, Excel, Powerpoint, Outlook)
- Easy definition of responsibilities, roles, sites, countries (from master data)
- Modification of documents exclusively by authorized persons
- Full master data management
Document creation and control
- Best-practice workflows, e.g., for editing, review and approval processes
- Electronic signatures according to 21 CFR Part 11
- Workflow-based creation, review, and approval of documents
- Traceable versioning of all documents in the system (version history)
- Generation of new versions exclusively on the basis of released/valid versions
- Automatic status assignment for documents
- Control via predefined workflows; intuitive definition of workflow participants
- Definition and assignment of tasks in document workflows
- Notifications when tasks are assigned or completed
- Possibility of serial or parallel review and approval cycles
- Absence assistant and deputy regulation
- Study lifecycle management Status management
- Automatic status changes for documents (“in process”, “for review”, “for release”)
- Automated version management for documents, including overriding and archiving of previous versions
Publishing
- Automatic conversion of documents to PDF
- Real-time publication
- Target-group oriented information distribution for new or changed documents
- Distribution for information or acknowledgement (to groups or individuals)
- Simple customer-specific modeling and configuration of release workflows
- Task distribution in personal task box and by mail (incl. link to document)
- Option to integrate DHC VISION training management
Verification
- Predefined workflows for document invalidation, invalidation, validity extension, reactivation, and archiving
- Document control by date and time period; cyclical resubmission of documents for review
- Manual, document-specific definition of the date for resubmission
- Direct review run or validity extension from the resubmission process
- Reminder and escalation mechanisms to meet deadlines (lead times)
- Complete reference list for documents, incl. indication of their context of use
- Support of document invalidation by authorized persons
- Dynamic display and simple processing of proposals for corrections/modifictions
- Editing / revision of new versions parallel to the use of valid versions
Analytics
- Number of documents per status and type
- Overdue management: validity of documents, incl. status information
- Drill-down to documents with status information
- Display of essential document attributes or properties
- Quick view of the individual document incl. accompanying documents
Reporting
- Highly performant cognitive search
- Comprehensive search mechanisms and filter/selection functions
- Integrated full text search of document content and attributes
- Wide range of selection parameters to support a broad range of use cases
- Configurable status reports and resubmission schedules
- Automated monitoring of document validity
- Reminder/escalation mechanisms for monitoring documents with limited validity
- Comprehensive mechanisms for status tracking (tracebility)
- Workflow monitoring
Attachments and appendices
- Creation of documents/information as attachments or additional information
- Reference to further documents, e.g., checklists, scans, incl. multiple selection
- Links/references to additional objects (processes, sites, persons, countries, etc.)
- Targeted modification of additional information/references without influencing the main document
- Automatic version and status management
- Intelligent notification system
Events, Notifications, Communication
- Notification Event Modeling Framework for automated, accurate and timely notification of people, roles/groups or systems.
- Flexible and appealing design of notifications (including HTML); multilingual notifications; to different internal or external (e.g., sites) addressees
- Documentation of notifications in the audit trail
- Full traceability of information events and content
Security
- Audit-proof storage and management of documents in freely definable structures
- Complete version and change history (audit trail)
- Write protection of already signed or released documents
- 21 CFR Part 11 compliance (electronic records, electronic signatures)
- Fine-grained, differentiated role and authorization concept for studies and study documents
- Predefined roles, profiles, user groups, and authorizations
- Compliance with the highest security requirements
Integration and connectivity
- Tight integration of eTMF and overarching digital compliance processes
- Connection of eTMF to the powerful document management in DHC VISION
- Combination of study documentation and training in DHC VISION training management
- Reliable handling of deviations through integration of eTMF and deviation management
- Efficient management of changes in DHC VISION Change Management
Validation and compliance consistently in view
DHC VISION is specially designed for use in highly regulated industries. The solution fulfills the GxP guidelines and directives of the FDA, EMA, PIC/S or ICH, as well as 21 CFR Part 11, both technologically and technically (business processes). The Validation Package is available for validating the system. Consisting of the Validation Accelerators (complete documentation set for validation) and the Validation Services for adapting the documentation to your individual situation.
Matching products

SOP CONTROL
The optimal solution for digital management and control of your specification documentation. Secure, controlled, traceable and compliant (including 21 CFR Part 11).

TRAINING
The perfect and seamlessly integrable addition to SOP management. Digital processes set new standards in “Training Compliance”.

LEARNING SUCCESS
If “Read and Understood” is not enough, successfull learning is checked at the end of the SOP Closed Loop processes.

CAPA
Automated control of Corrective and Preventive Actions. No action will be forgotten until it is fully implemented.

DEVIATION
Systematic and controlled documentation, analysis and resolution of non-conformancies to improve quality and compliance.

COMPLAINTS
Whether they come from costumers, suppliers or internal processes – complaints are maneged in a controlled manner. This is done with the highest degree of regulatory (process) security.

CHANGE
System processes for Change Control ensure precise management and communication of changes of any kind, from the beginning to the end.

AUDIT
Indispensable for an integrated management system. An entirely digital and user-friendly audit process. The ideal addition to the other quality modules.
Your information package
Get an impression of this and other products or read what insights we have gained from research and development. Take advantage of our exclusive content such as white papers or study results on the digitization of quality and compliance processes. Put together your desired media easily and conveniently.
Worth knowing | News | Latest

RegTech Insights: Can artificial intelligence improve post-market surveillance? An industry survey is to provide answers.
Post-market surveillance is a mandatory task for manufacturers of medical devices and involves a great deal of effort. Whether and how...

Verla-Pharm modernizes regulatory compliance with eQMS solutions from DHC Business Solutions
Verla-Pharm Arzneimittel GmbH & Co. KG relies on the industry and solution expertise of DHC Business Solutions for the modernization...

Synergy and efficiency gains in quality management at Corden BioChem with DHC VISION
Corden BioChem GmbH, located in Höchst near Frankfurt, is one of the world's largest industrial production partners for biotechnologically...
FAQ
What is a Trial Master File?
With the discussion about the approval of Corona vaccines, the term “clinical trial” is known to the general public. A clinical trial tests the performance, efficacy and safety of medical devices, forms of treatment, active substances, etc. and is conducted with patients or volunteers.
The Trial Master File (TMF) is a collection of documents from a clinical trial. The TMF summarizes the essential files of the study while ensuring the integrity of the clinical data and compliance to Good Clinical Practice (GCP).
In a clinical trial, the following actors usually work together: The research company (sponsor), which has its product tested for performance, efficacy and tolerability; a contract research organization (CRO), which conducts the study on behalf of the sponsor; and one or more clinical institutions (investigators), which conduct the trial according to applicable standards and in accordance with the study plan. One TMF is created per Clinical Trial.
Which regulations and standards are relevant for a TMF?
The GCP – Good Clinical Practice – defines an internationally recognized standard for planning, conducting, monitoring/auditing, documenting, evaluating, and reporting clinical trials, based on ethical and scientific aspects. The content of a TMF is regulated in the EU Directive 2005/28/EC.
Relevant regulations of the European Medicines Agency (EMA) are: Guideline on the content, management and archiving of the clinical trial master file (paper and/or electronic), (EMA/INS/GCP/856758/2018); Guideline for good clinical practice E6(R2), Step 2b (EMA/CHMP/ICH/135/1995); Guideline for good clinical practice E6(R2), Step 5 (EMA/CHMP/ICH/135/1995).
The structure of a clinical trial and the corresponding documentation is defined in the Drug Information Association (DIA) reference model TMF.
What are challenges associated with TMFs? Does digitization help?
The particular challenges with TMFs are:
- the number of people/organizations involved (sponsor, CRO, investigators) and their different tasks and interests;
- the multitude and heterogeneity of data and documents, their completeness, retrievability, availability and auditable storage or documentation according to regulations in place;
- media discontinuities between analog, partly handwritten, data on the one hand and digital data on the other; the challenge is to manage such heterogeneous documents in a consolidated way due to the low degree of digitization, among other things;
- the lack of integration of IT systems on the sponsor’s, CRO’s and investigators’ sites, i.e., the heterogeneous and redundant IT-infrastructure, which impede the continuous exchange of data.
This makes it difficult to conduct or document a study in a way that can be audited by regulatory and licensing authorities.
Audit reports from regulatory authorities, therefore, regularly complain about the management of TMF and the comprehensible documentation of studies according to compliance regulations. Digital processes and appropriate AI support in an eTMF may help making clinical trial documentation more reliable and compliant. The research project NextGenTMF is working on a solution under the leadership of DHC. More information is available here.

